清华AI找药登Science量子位
一天筛选十万亿次,中国AI找药又有新突破!
清华大学智能产业研究院(AIR)联合清华大学生命学院、清华大学化学系在Science上发表论文:《深度对比学习实现基因组级别药物虚拟筛选》。
团队研发了一个AI驱动的超高通量药物虚拟筛选平台DrugCLIP。
DrugCLIP能让AI从海量化学分子里,迅速筛出那些最有希望和疾病相关蛋白结合的“候选药物分子”。
24小时内,DrugCLIP能完成10万亿次蛋白–分子配对计算。
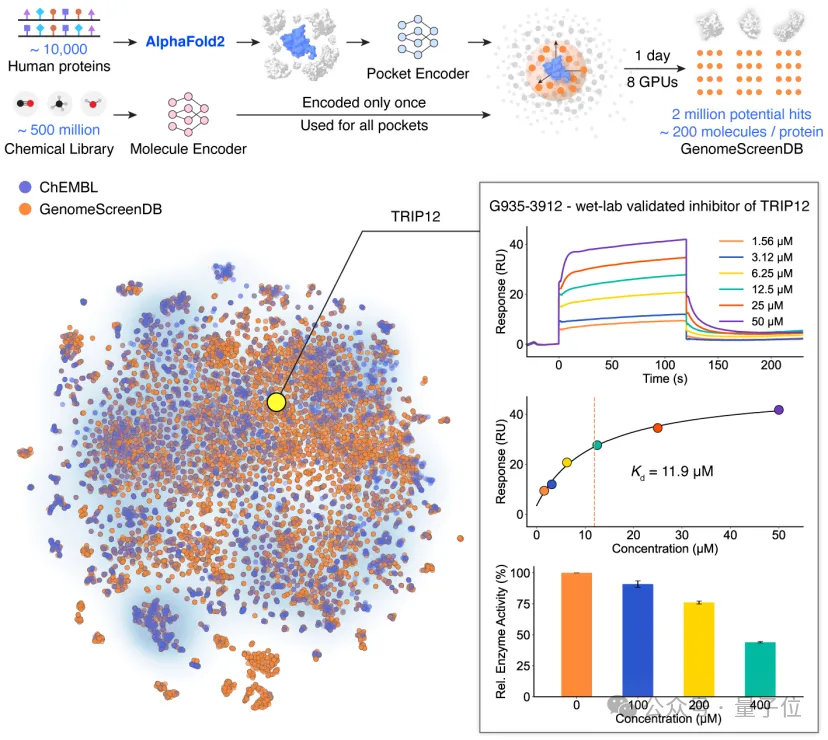
依托该平台筛选,团队打通了从AlphaFold结构预测到药物发现的关键通道,不仅为抑郁症、癌症、帕金森等疾病筛选出了潜在药物分子,还首次完成了覆盖人类基因组规模的药物虚拟筛选。
目前,相关数据已经全部对外开放。
90%的蛋白靶点难找药
过去药物筛选的难点,主要集中在三点上,一是慢,二是无从下手,三是范围太窄。
先看一个背景数字。
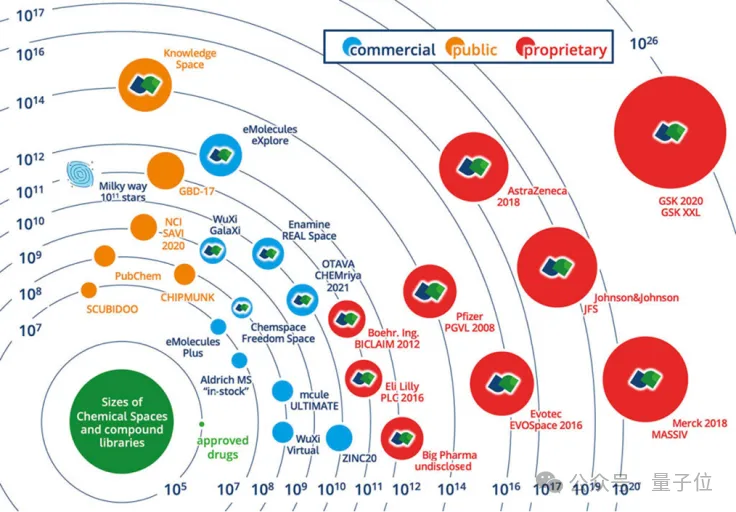
人体内大约有2万个编码蛋白质的基因,其中的相当一部分与癌症、抑郁症、神经退行性疾病密切相关。
但现实是,目前真正拥有成熟药物的蛋白靶点,只占其中10%,剩下的90%,还没找到药。
△化学空间大小示意图(引用:Gastreich, M. BioSolveITDrugSpace2022)
第一个原因,慢。
传统的筛选方法,比如分子对接,需要逐一计算“这个分子能不能和这个蛋白结合”,一次评估虽然只要几秒钟或几分钟,但在现实情况下,以筛选1万个蛋白质靶点、每个靶点面对10⁹个候选分子为例,需完成约10¹³次蛋白-配体打分。
即便使用当前最先进的分子对接工具,也得需要2亿CPU天。
第二个原因,无从下手。
很多疾病相关蛋白根本没有实验测出来的三维结构,传统方法无从下手。
而且在真实世界里,没用的分子还远比有用的分子多,这些好分子容易被埋没在噪声里。
第三个,范围太窄。
算力成本摆在这儿,只能围绕热门靶点筛,工作很难在全基因组的尺度上推进。
不过,DrugCLIP正是冲着这三点来的。
给蛋白和分子画像
先概括一下它的方法,就是先教会AI为目标进行画像,捕捉其结构神韵,再做极速配对。
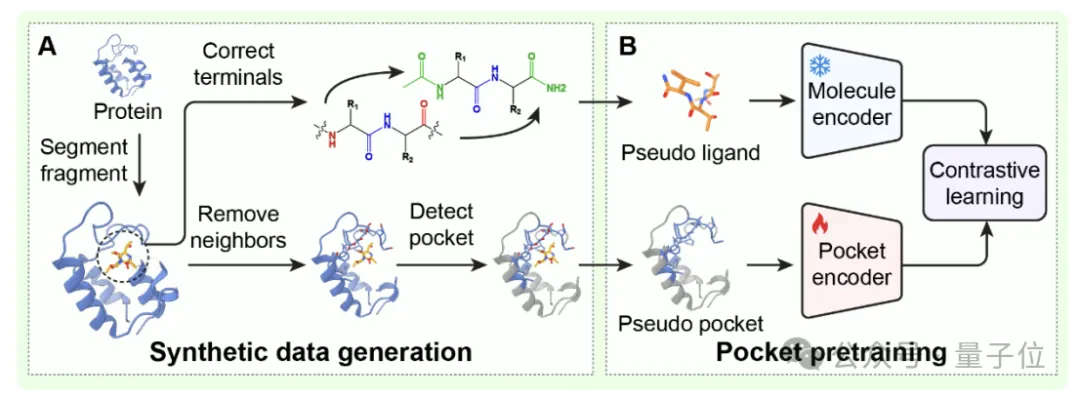
研究者用对比学习训练了两个AI编码器。
一个给蛋白质上的结合口袋画像,另一个给化学分子画像。
“结合口袋”是指蛋白质表面能够与小分子结合的特定区域,这里的“画像”是指生成特征向量。
训练时,AI会被明确告知:能结合的一对儿,画像要尽量接近,即对应的特征向量要尽可能相似;不能结合的,画像要尽量拉远。
这样一来,AI就能逐渐学习并掌握蛋白质与分子之间的结合规律。
为了让模型从一开始就领悟这种结构神韵,团队设计了一套创新性的预训练策略。
他们从已有的蛋白质结构数据中,切割出短片段模拟成“假分子”,同时将周围区域当作“假口袋”,一次性构造出了550万组训练样本。
在这套练手数据上打好基础后,再用真实的蛋白-分子数据进行微调,保证了泛化能力和精度。
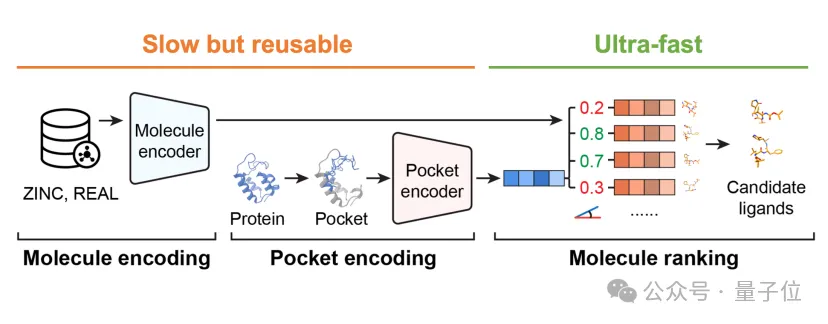
模型训练完成后,真正的筛选过程就变得简单高效了。
DrugCLIP创新性地将传统基于物理对接的筛选流程转化为高效的向量检索问题。
研究者先把5亿个候选分子全部画像完存起来,当遇到一个新的蛋白口袋时,只需要给它生成一个向量表示,再和所有的分子算相似度、排个名,排在前面的就是最有希望的候选分子。
该模型结合对比学习、3D结构预训练与多模态编码技术,能在三维结构层面精准建模蛋白-配体间的相互作用。
训练后的高潜力分子将自然聚集于目标蛋白口袋的向量邻域,能够有效支撑快速的大规模虚拟筛选。
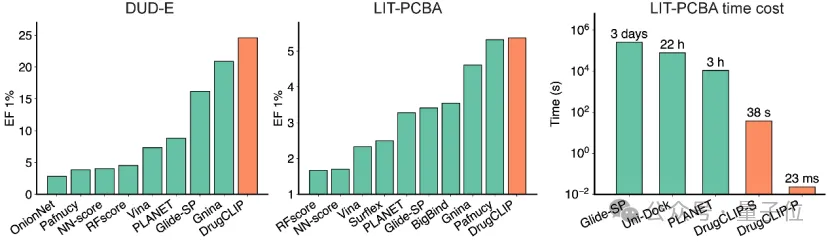
依托这一机制,DrugCLIP在128核CPU+8张GPU的计算节点上日处理能力达10万亿次,对比传统方法实现了百万倍提升。
首次完成了人类基因组规模的虚拟筛选
速度之外,更关键的是它真能找到有用的分子。
在标准的虚拟筛选基准测试DUD-E、LIT-PCBA中,DrugCLIP在把有效分子从大量无效分子中提前筛出来这件事上,明显优于传统分子对接工具和多种已有AI方法。
并且在LIT-PCBA数据集上筛选速度远超其他方法。
而且它对结构误差、陌生蛋白家族、从未见过的分子类型都表现得相当稳定,没有出现“一换场景就失灵”的问题。
实验室验证结果也让人眼前一亮。
以抑郁症相关蛋白为例,研究者从筛选出的78个分子里,找到8个能激活这个蛋白的“激动剂”。
其中最好的一个分子,和蛋白的结合能力达到21nM(数值越小结合越强,100nM以下就是优秀水平),在细胞系中也有显著活性。